

There are various regulatory pathways that MedTech companies may need to follow when seeking premarket review from FDA, but determining the best path for your device involves many moving parts, resulting in a complex situation for consideration. Two of the most common and well-known pathways are the 510(k), or the Premarket Notification, and the Premarket Approval (PMA).
Despite often being compared as similar submission types, the two pathways are significantly different from one another. Each route must be followed under different circumstances, and each one has its own timeline, submission requirements, and submission contents. Let's review each pathway and dive into the main differences between the submission types.

Table of Contents:
- Medical device classifications and FDA submissions
- What is a 510(k), and is this the right path for your device?
- What is a PMA, and is this the right path for your device?
- Let’s Review – what is the difference between the 510(k) and PMA submission types?
- How CROs can help with your medical device pathway
Medical device classifications and FDA submissions
There are three main risk classes of devices in the United States – Class I, II, and III. Each class has regulatory pathways that are most applicable based on the amount of review FDA has determined to be appropriate for devices of each type. Class I devices are often 510(K) exempt, with some exceptions for devices that still require 510(k). Many Class II devices require a 510(k), but some may be exempt from premarket submission requirements or sometimes, for novel technologies, may require a De Novo. Lastly, almost all Class III devices require a PMA, though the occasional Class III 510(k) exists.
However, before we dive further into the differences between 510(k)s and PMAs, let’s briefly discuss the De Novo submission process, which is the middle ground between the two pathways. You would pursue the De Novo pathway if you had a device with a moderate risk level but with no suitable predicate device. For more information on predicate devices, read the article section below, What is 510(k), and is this the right path for your device.
Instead of requiring a PMA, FDA established the De Novo pathway to provide a thorough review of these novel devices without necessitating the same level of requirements as a PMA.

Understanding the classification for your device is necessary in understanding the ultimate submission pathway for your product. To get a feel for how you might be classified, you can search the FDA classification database and find a product similar to your device to determine your medical device class. However, to appropriately classify your device, you will need to establish and evaluate your device's intended use and indications for use with respect to the established regulations.
Certain claims may result in different product codes, classifications, and/or regulatory pathways, which may mean different regulatory submission requirements for your device. Because this can get complicated, companies are advised to work with a consultant or someone well-versed in regulatory affairs to explore options available to your company and its device.
Consultants or contract research organizations, like Proxima, can help ensure you meet the requirements for a 510(k) or PMA submission and speak with FDA on your behalf during your pre-market submission process as well as through earlier interactions, like Pre-Submissions (Q-Subs). Watch a quick overview from our regulatory team for more information on Pre-Submission meetings.
Overview of Medical Device Class Determination, Submissions Associated with Each Risk Level & Average Timeframe
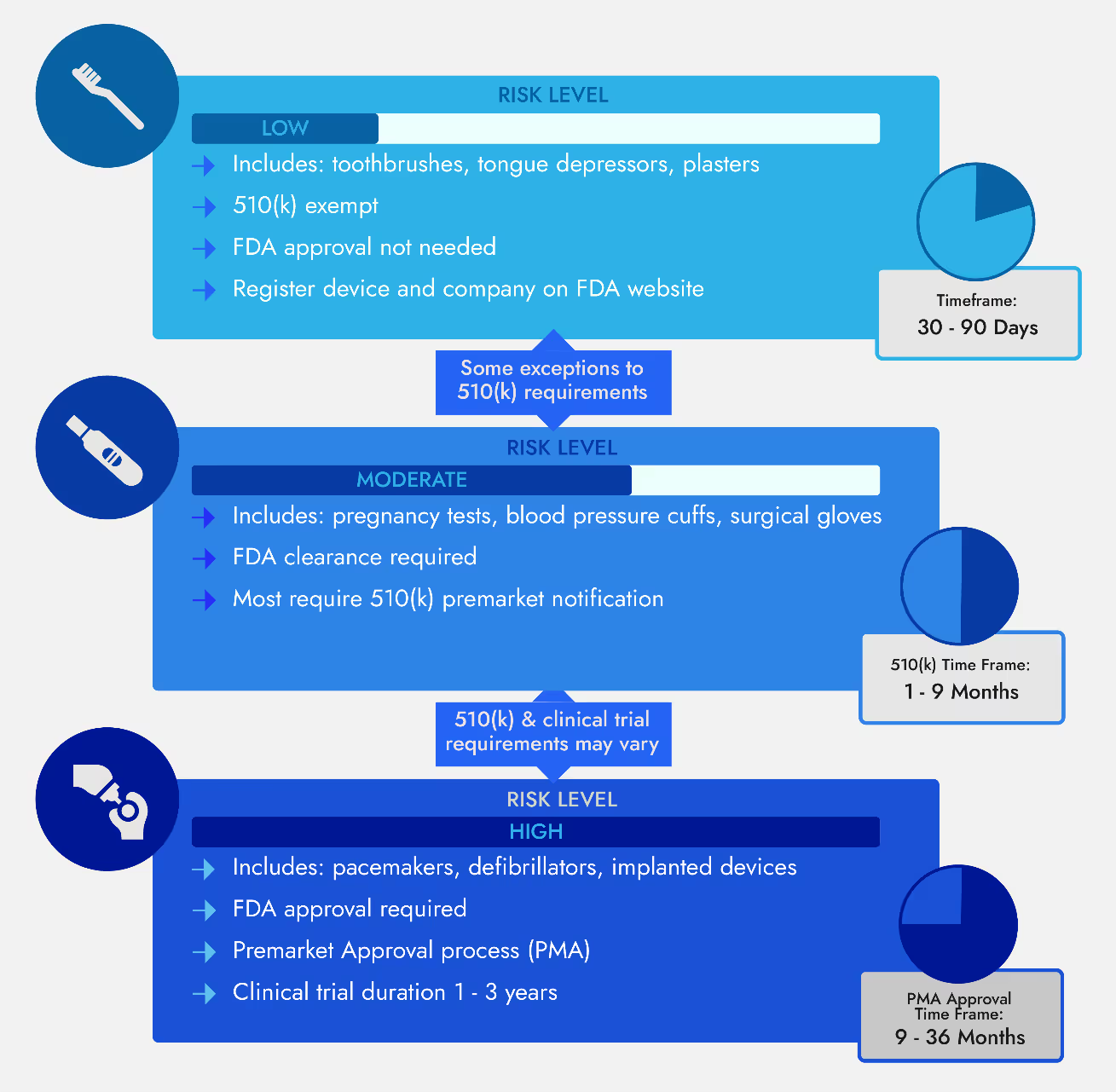
So, if you’re looking at a product with what you think might be two possible regulatory pathways—a 510(k) or a PMA, how do you determine which applies?
You’ll first need to know a bit about the two pathways.
What is a 510(k), and is this the right path for your device?
510(k) Notification
A 510(k) is a premarket submission for Class II medical devices that can show the device is substantially equivalent (SE) to one or more products that are currently being sold legally in the U.S. with the same intended application and comparable technological features. This legally marketed device is also known as the ‘predicate device.’ After successfully receiving an FDA determination of substantial equivalence through a 510(k), a device would be recognized as having FDA clearance and may be legally marketed in the US.
There are several 510(k) submissions types to choose from. However, if this is your first cleared product, you are most likely to go with Traditional 510(k) (#1), below. The Abbreviated 510(k) (#2) is seldom used, and Special 510(k) (#3) is only applicable if you already have a product on the market:
- Traditional 510(k) - Used for any original 510(k) or new submission for a novel device that utilizes an appropriate predicate device to demonstrate substantial equivalence
- Abbreviated 510(k) - Used for submissions that rely on voluntary consensus standards, FDA guideline materials, and proof of compliance
- Special 510(k) - Used when a manufacturer modifies its own legally marketed product that has already been through a 510(k), provided that the adjustment does not significantly change the product's technology or intended use
For a Traditional 510(k), within 60 days of filing, you may receive a request for additional information, also called an AINN. You must respond to the deficiencies cited in the AINN letter within 180 calendar days. This means, no matter how big the issue, you have approximately 6 months to address it. During the 180 days, you can solicit FDA feedback on your approach to addressing the deficiencies through Submission Issue Requests.
At the end of the 180 days, you must submit your response. The FDA will then have an additional 30 days or so to review, rendering their final review timeline of 90 days. The submission will then be published in the 510(k) Premarket Notification database if it is cleared by the FDA.
What is a PMA, and is this the right path for your device?
Pre-Market Approval (PMA)
PMAs are rigorous regulatory submissions that must be provided to the FDA to demonstrate the efficacy and safety of a Class III medical device. Devices that have received approval after submission of a PMA would be recognized as FDA-approved. For more information about PMA regulations and its approval processes, read this Proxima article, Navigating the Pre-Market Approval Application Process for Your Class III Medical Device.

There are also several PMA submissions types. Of the types, if this is your first (or original) PMA, you are likely to select either Traditional PMA (#1) or Modular PMA (#2). PMA Supplements (#3) are for modifications to your device when it’s already on the market.
- Traditional PMA - In this submission format, all required contents are submitted in a single PMA application. Manufacturers who have finished all of their clinical trials may use this approach
- Modular PMA - In this format, PMAs are divided into modules, which are then submitted over time to complete a comprehensive PMA application. Most medical device firms are advised to follow the Modular PMA path because it enables businesses to finish sections, or modules, over time while clinical studies are still being conducted and analyzed
- PMA Supplement - Required for modifications to a device for which the applicant has an approved PMA
Due to the more stringent and robust elements required for a PMA submission, FDA will have 180 total days to review the application, compared to the 90 days for a 510(k) review. However, the timeline often takes much longer due to FDA requests for additional information from the applicant and the potential for FDA to request an Advisory Panel. After a thorough review process of the clinical data and all supporting information, you will receive an order either approving or rejecting your medical device for use.
Let’s Review – What is the difference between the 510(k) and PMA submission types?
The PMA process differs significantly from the 510(k) approach in that it is more demanding and in-depth. While a 510(k) only requires a demonstration of substantial equivalence to the chosen predicate, PMAs require a full dossier of evidence to support the safety and effectiveness, usually including large multicenter clinical trials. In many cases, even if a 510(k) requires clinical data, the necessary trials may be smaller cohorts than that required for a PMA. Furthermore, PMAs require quality management audits while 510(k)s do not.
While you may be wondering why anyone would pursue a PMA when 510(k) is clearly the easier pathway, there are certain benefits. If you receive approval for your device through a PMA, no one will be able to use your device as a predicate in a 510(k). This means that any other similar devices will need to go through the PMA approval process as well, which means higher barriers of entry to the market, resulting in potentially fewer competitors. Because a PMA has such stringent data requirements, you will be able to show the value of your device to the market in a much more robust manner than most 510(k) devices.

How CROs can help with your medical device pathway
Preparing a 510(k) or a PMA presents unique difficulties, so it is essential to get advice from professionals who have experience with numerous application types and are familiar with FDA regulations.
In addition to assisting you with the Pre-Submission process, CROs, like Proxima Clinical Research, also offer regulatory assessments and regulatory strategies to help you determine the class of your device and its clinical and submission requirements.
- Regulatory Assessments - Data is gathered on the relevant medical device to determine the class and pathway that innovative medical device companies should take to market
- Regulatory Strategies - Similar to a regulatory assessment, but with more in-depth research and presentation to yield all-inclusive feedback on the testing requirements for your medical device
Employing a CRO during the 510(k) or PMA submission process enables you to audit and assess progress throughout the FDA approval process, quickly gather additional feedback from the FDA when questions or concerns occur, create deliverables to stay on schedule, and set work plans and goals. We are here to support you, so you won't have to take this path alone.
.avif)

