

The Premarket Approval process can be a demanding and expensive pathway for businesses.
If you think you might be a Class III medical device, knowing this process is imperative for you and your organization.
There is a lot to unpack in preparing for a PMA, so companies like Proxima exist to help you strategize and perform. As a regulatory consulting and clinical trial execution firm that ensures MedTech companies and their products adhere to all regulatory standards and developmental life cycles, Proxima is ready to take the pain out of your PMA. Visit us at www.proximacro.com for additional information on the PMA submission process.

Table of Contents:
- What Premarket Approval (PMA) is and how it came to be
- Types of PMAs
- What to do before submitting a PMA
- What to submit for Premarket Approval
- The PMA FDA Review Process
- How to reduce PMA regulatory costs
- PMA Clinical Studies
What Premarket Approval (PMA) is and how it came to be
After Congress passed the Medical Device Amendment in 1976, the Food and Drug Administration (FDA) assembled a panel to decide classes based on risks associated with the device type. As a result, a three tiered system was developed, separating devices into Class I, Class II, and Class III.

Per the Amendment, lower-risk devices require a 510(k) or are labeled as exempt from pre-market notification (find out more about 510(k) exemptions here), and Class III devices require evaluation by FDA before being placed on the market. Thus, the Premarket Approval (PMA) pathway was born.
For PMAs, the review process requires applicants to provide sufficient evidence of well-controlled bench, animal, and clinical testing results in addition to having a manufacturing audit.
How does this differ from other submission types?

So, how can you assess if your device needs a PMA?
Let's examine the characteristics that define a Class III device:
What is a Class III device
- Devices that support or sustain human life
- Devices that play a vital role in preventing the impairment of human health
- Devices that are implanted
- Devices that present a potential risk of injury or illness.
Besides classifying device types, there are also different submissions types for each class of device.
But just to be sure you understand the various FDA classifications, take a second to learn more about the classifications with a Proxima original, How the FDA Classifies Medical Devices. Check it out and come back here to learn more about the different types of PMAs.

Ok, now that you’re up to speed on the types of classifications, there are also sub-types of PMA applications to choose from as well. (You didn’t think it’d be that easy, did you?)
Types of PMAs
Depending on the kind of permission being sought, there are several PMA applications to choose from. However, the amount of time, effort, and money saved can vary depending on the program you select.
- All required contents are submitted in a single PMA application. This method is best for manufacturers that have completed all their clinical testing
- PMAs are broken down into modules and submitted over time, completing a full PMA application when combined. The Modular PMA route is recommended for most medical device entities because it allows companies to complete sections or modules, and submit them to FDA while clinical studies are still in progress
- Required for a change affecting the safety of a device for which the applicant has an approved PMA
- Includes all additional submissions to a PMA or PMA Supplement before approval or all correspondence after approval
Now that you’ve established that you might be submitting a PMA and what type of PMA you’re likely submitting, what’s next?
What to do before submitting a PMA?
Before filing a PMA, it’s good (and necessary) to get in touch with the FDA via the Q-Submission process. In addition to building a rapport with the FDA review team, this early connection enables the sponsor to get advice on the device's development and testing plans.
(As a bonus: By identifying all tasks required for final approval, you will be validating your testing plan, which can encourage additional investment.)
As a part of the pre-submission process, you will have the opportunity to have a teleconference with FDA to discuss specific queries and details on pre-clinical studies, IDE requirements, human participant studies, and pivotal clinical trials.
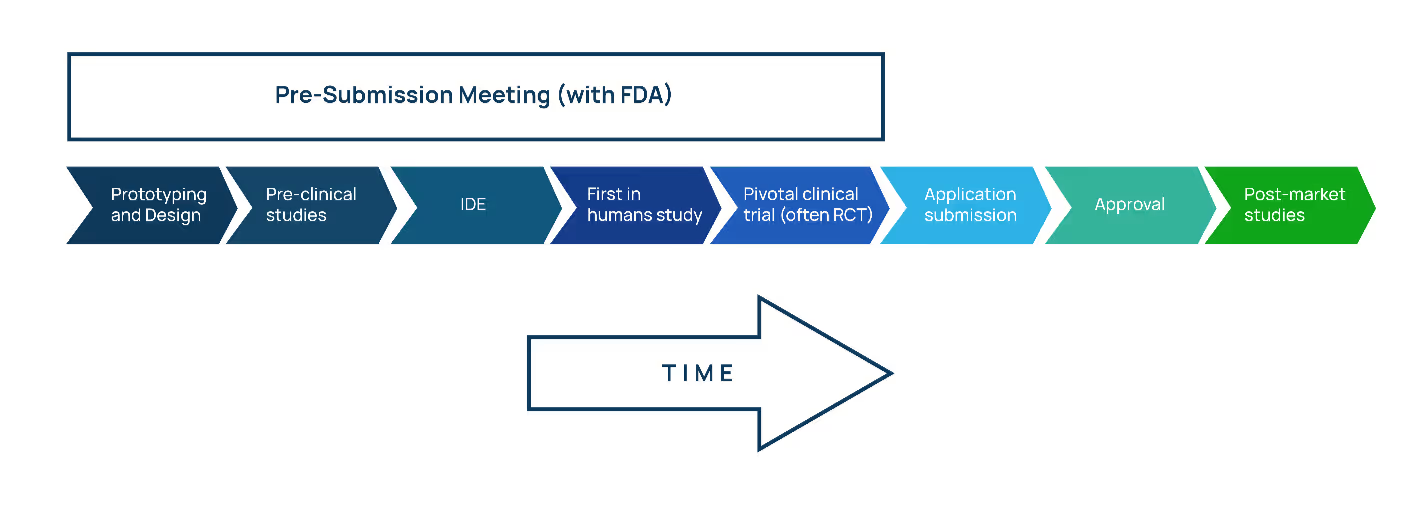
Listen to Proxima’s Q-Submission Program video to learn more about this meeting and the advantages it might provide for your medical product.
Now that you’ve had your pre-sub (or pre-subs, more likely) and you’ve completed your testing, you’re now ready to submit!
This takes us to the next step…
What to submit for Premarket Approval
For a complete PMA submission, the FDA requires several elements:
- A cover letter indicating if the PMA submission is a Traditional PMA, Modular PMA, PMA Supplement, or PMA Amendment, and a table of contents including:
- Separate sections for nonclinical lab studies and clinical human studies
- Identification of trade secrets, confidential or financial information
- A thorough summary of the submission, including:
- The purpose of the device and the patient population, disease, or condition the device is meant to cure or mitigate
- Device description; its function, name, and manufacturing process
- Other commercially available devices like the device being submitted
- A summary list of foreign and U.S. countries where a device, like the one being submitted, has been marketed and withdrawn
- Summary of results obtained from nonclinical and clinical studies
- Conclusions from the study data showing the device is safe for use as described in 21 CFR 860.7
- A complete description of the device, including pictures, all its functional parts, properties of the device relating to its treatment, how it works, and its manufacturing, packaging, and storage processes
- Discuss what performance standards or voluntary standards the device meets or deviates from
- A technical section with sufficient data that includes:
- Results of nonclinical laboratory studies
- Results of clinical studies on human subjects
- State if the study was compliant with 21 CFR 56, 21 CFR 56.104, 21 CFR 56.105, 21 CFR 50, or 21 CFR 812; if not compliant, state a brief reason for noncompliance
- If data are represented by only one investigator, include a justification as to why a single investigator is sufficient to demonstrate the safety of the device
- Bibliography of all published reports on the safety and effectiveness of the device
- If practical and if requested by the FDA, a sample of the device and its components should be sent for testing. If impractical, the applicant should provide the FDA with a location at which the device can be examined and tested
- Copies of all proposed labeling for the device
- An environmental assessment and environmental impact statement per 21 CFR 25. However, disregard if the device is not the same type and for the same indication as a previously approved device
- Financial certification or disclosure statement
If the PMA application lacks elements and critical results or is incomplete and inconsistent, the FDA may refuse to file or delay the PMA application for further review. Read more about the requirements of a PMA submission to get a better understanding of what to do when information is left out or when new data is added to the PMA.
Again, we know, this is a lot of information and a bit complicated, which is why we are here. It’s not that you couldn’t handle it if you had to, but why should you? You’ve got more important things to worry about. As consulting experts, Proxima simplifies this process by making sure the PMA application is accurate and complete. Proxima drafts, compiles, reviews, and submits the application to then interface with the FDA throughout the process to put medical device manufacturers at a distinctive advantage from any risks of delays.
And now that you (or we) have submitted your application, what happens to your valuable file?
The PMA FDA Review Process
There is a four-step review process for evaluating the PMA application. The Acceptance and Filing Review is the first step.
If the PMA application is filed and accepted, a 180-day Substantive review of the PMA will begin on the date of filing. Applicants may submit a meeting request no later than 70 days after filing to schedule a Day-100 Meeting and discuss the status of the application review.
After the Day-100 meeting, the FDA will continue to communicate with the applicant at least every 4 weeks until the review has been completed.
The next few steps include the Panel Review and Final Deliberation. The Panel Review is meant to take place with experts reviewing the PMA for further recommendations. During this review, the advisory committee may reach out to applicants with questions and requests for additional information. The panel is meant to provide advice on PMA submissions and regulatory issues.
If the application is rejected from the initial Acceptance and Filing review process, the applicant may submit a written request within 10 business days of receiving the refusal to hold a meeting with the Office of Health Technology (OHT) and review the outcome.
After the review process is complete, a final decision on the PMA must be made. One of the following Approval or Denial orders may be issued by the FDA:
- Approval Order
- You will be issued an approval order and the public will be notified online through a Summary of Safety and Effectiveness Data (SSED) document
- Approval Letter
- You will be issued an approval letter if the FDA believes the PMA can be approved after additional information can be submitted or certain conditions can be met
- Not Approval Letter
- If there is a lack of significant information in the PMA, the FDA may issue a not approval letter
- Denial of Approval
- If you go against any PMA regulations, for example, including false statements or denying facility and control inspections, a denial of approval may be presented
- Withdrawal of Approval
- If you fail to follow PMA regulations, if post-approval requirements have not been met, or if laboratory studies haven’t been conducted in compliance, an opportunity for a hearing will be provided before issuing an order withdrawing approval
- Temporary Suspension
- If an opportunity for a hearing was conducted after being presented with a Withdrawal of Approval Order, the FDA will then decide if continuing with this device would cause adverse health concerns. If so, the FDA will suspend the PMA.
The progress of the device depends on your response to each Approval or Denial order. You have the option to withdraw the PMA, make a request for reconsideration, abide by the FDA and provide additional information, or consent to post-approval research depending on the order letter received.
From early-stage advising consultations to all-inclusive clinical trial services, Proxima works with businesses throughout this developmental phase to ensure attention is given to the expected results. For a graphic summary, learn more about the PMA FDA submission, review, and notification cycle below in detail.

And, as a bonus, here are some tips to help with reducing the cost incurred by the regulatory process:
How to reduce PMA regulatory costs
- For innovative devices, the Breakthrough Device Program exists to allow for more frequent interaction and communication, which may reduce the time and costs from the developmental phase through the marketing decisions
- PMA submission fee waivers are included for first-time businesses with gross receipts of sales of no more than $30 Million
- PMA submission fees are exempt if the applicant is from a State or Federal entity, not distributing commercially
- PMA submission fees are exempt if the device is intended solely for pediatric use
- Applicants can determine whether they qualify for the Small Business Determination Program to pay a Small Business fee rather than the Standard Fee. Make sure to submit it in a timely manner, as it can take up to 60 days to process.

PMA Clinical Studies
Having a well-controlled clinical study that complies with Good Clinical Practice regulations is necessary for the quality and acceptance of the evidence. Before starting the PMA process, learn more about the FDA approval criteria for clinical studies and get comfortable.
As a contract research organization that provides clinical trial services and consultation for medical device companies, Proxima Clinical Research is an inviting force for businesses like you looking to overtake this safe route in bringing your medical device into the market.
.avif)

